Only a relatively small fraction (about 10-15%) of all Parkinson’s cases can currently be explained by genetic causes. As a result, if you have Parkinson’s disease, your children are not necessarily at any significantly increased risk for getting the disease. There are exceptions, however, where Parkinson’s disease does seem to ‘run in the family‘, and with the revolutionary advances in the ability to sequence human genes, starting towards the end of the 20th century, it has been possible to figure out why. The insights gained are relevant not just to the patients who have those mutations but to all people with Parkinson’s. Here, I will focus only on genes that have been shown to cause Parkinson’s; there are a number of other genes that have been shown to increase the risk of developing Parkinson’s by varying amounts. Both types of genes are potential targets for Parkinson’s drug discovery efforts.
The first genetic cause identified for Parkinson’s disease is alpha-synuclein. This protein had previously been known as a major component of Lewy bodies, which are identified in the brains of most deceased people who had Parkinson’s disease. Because this protein plays such a big role in our understanding of Parkinson’s, I discuss it separately in some detail. Only a very small fraction of Parkinson’s cases can be explained by abnormalities in this gene, however.
Subsequently, quite a few other genetic causes of Parkinson’s have been identified, and interestingly, several of these genes are involved in a particular ‘garbage disposal’ function in the cell, the autophagy-lysosomal system. Autophagy is a process for eliminating unneeded or defective cellular components, which ends with the components being delivered to the lysosome to be broken down and reused. In Parkinson’s disease, autophagy is of particular interest with respect to degrading defective mitochondria (‘mitophagy’) and alpha-synuclein aggregates, since both of these have been implicated in the death of dopaminergic neurons. Also recall that the few chemicals known to cause Parkinson’s do so by damaging mitochondria.
Here, I will only provide a brief summary of various important genes associated with Parkinson’s, and as time permits, I’ll add more detailed information on some of them in separate pages.
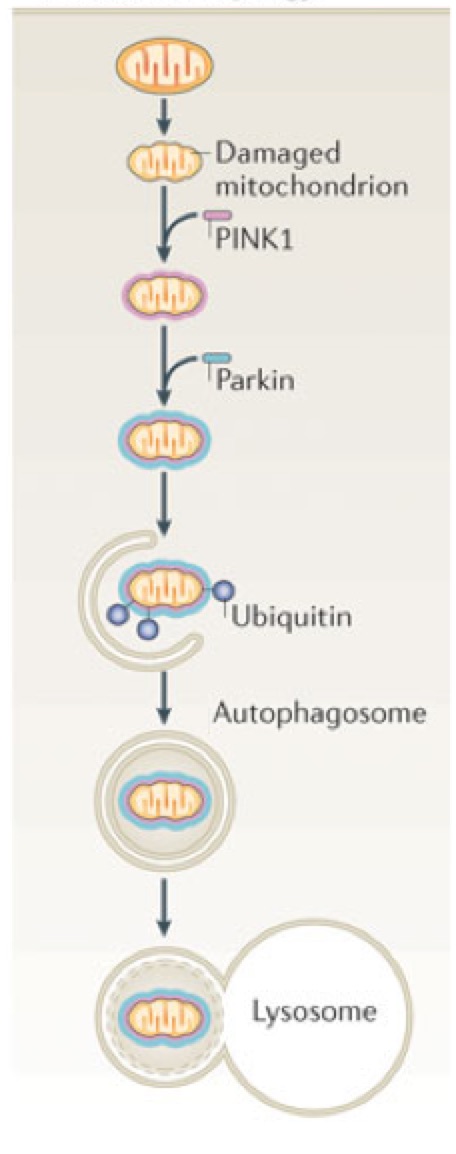
Mitophagy, a mechanism for recycling damaged mitochondria, may be impaired in Parkinson’s disease. Mutations in the PINK1 and Parkin proteins have both been shown to cause the disease. Reprinted and slightly adapted by permission from Macmillan Publishers Ltd: Nature Reviews Molecular Cell Biology (Youle and Narendra, Mechanisms of Mitophagy), copyright 2011.
- Parkin (PARK2): A “ubiquitin ligase” that attaches ubiquitin to damaged mitochondria, an important step in initiating mitophagy. Ubiquitin is usually used to tag damaged or unneeded proteins for destruction by the proteasome, and Parkin seems to also perform this role for certain proteins. Parkin mutations found to cause Parkinson’s are frequently found in young patients.
- PINK1: Plays a key role in identifying damaged mitochondria and then recruiting Parkin to them. Colleagues at UCSF, led by Kevan Shokat, are doing interesting work aimed at rescuing defects in PINK1 as a potential Parkinson’s therapeutic strategy.
- Glucocereberosidase (GBA): A commonly mutated gene identified in Parkinson’s disease patients. Not exactly part of the autophagy or mitophagy pathways, but an important lysosomal enzyme involved in breaking down a particular type of lipid (fatty substance). Interestingly, mutations in GBA can also cause Gaucher’s Disease, which is clinically quite different from Parkinson’s, but also involves lysosomal dysfuction, and patients with Gaucher’s have significantly increased risk of Parkinson’s.
- Other genes associated with Parkinson’s, such as VPS35 and ATP13A2, also have connections to autophagy and the lysosome.
- DJ-1 (PARK7) remains relatively poorly understood but it seems clear that it is involved in sensing and responding to oxidative stress. Connections to other Parkinson’s genes remain relatively speculative, but oxidative stress does occur when mitochondria malfunction, and DJ-1 may play a role in detecting this.
- LRRK2 mutations are some of the most common known causes of Parkinson’s, particularly in certain populations such as Ashkenazi Jews. The protein encoded by the gene is complex and many aspects of the biology remain incompletely understood, although a number of studies (e.g., 1, 2, 3, 4) linked it to autophagy or Parkin. LRRK2 has been a major recent focus of Parkinson’s drug discovery (see, e.g., Pfizer’s efforts), although lung toxicity may present a challenge to further development of LRRK2 drugs.
- Even alpha-synuclein may play important roles in regulating mitochondria.
I don’t wish to imply that autophagy or mitophagy, or lysosomal function more broadly, are the only cellular functions disrupted in Parkinson’s disease; the biology of the disease is complex, and much remains poorly understood. But the genetic mutations causing Parkinson’s have provided important clues, many of which have pointed toward defects in the recycling function of lysosomes, and the autophagy and mitophagy pathways feeding into it, which ultimately can lead to the neuronal degeneration seen in Parkinson’s disease. Defects in lysosomes and autophagy are also linked to other neurodegenerative diseases such as Alzheimer’s.
